Amplicon Sequencing (16s, ITS)
Use amplicon sequencing to detect and classify bacteria and fungi




Amplicon sequencing services
We help you precisely and accurately to identify the microbial composition of your sample.
Using amplicon sequencing with ITS or 16s we can detect and classify the bacteria and fungi in samples like:
Agriculture samples
- plant
- root
- soil/dirt
- water/aqua
- bioreactor
Life science samples
- gut
- blood
- spit
- skin
- fecal/stool/feces
- biopsies
Targeted sequencing of a phylogenetic marker gene allows us to profile the taxonomic composition of a specific microbial kingdom, such as bacteria or fungi. In our amplicon sequencing services, we profile the bacterial microbiome by sequencing the 16S rRNA gene, and we profile the fungi microbiome by sequencing the internal transcribed spacer 2 (ITS2) sequence (the mycobiome).
A cost-effective approach
Amplicon sequencing is a cost-effective approach for profiling a specific part of the microbiome and can be used for most any sample – including soil, water, skin, and stool – to detect the different species found in the sample. In situations where there are expected to be relatively few microorganisms in the samples, amplicon sequencing has an advantage over shotgun sequencing as it targets the organism of interest and therefore does not use a lot of reagent to sequence other types of DNA in the sample.
Please see below for some more information about the 16S rRNA gene and the ITS sequence. These are the two marker regions most often purchased via our amplicon sequencing service.
Why chose amplicon sequencing?
- Allow specific profiling of the bacterial or fungal microbiome
- It is cheaper than shotgun sequencing although the cost difference continues to reduce, and mostly depends on the desired sequencing depth and the sample type.
- The data is easier to process and analyze, and data storage is fewer challenges due to small amounts of data.
- It has some advantages over shotgun sequencing for samples with low microbiome concentration.
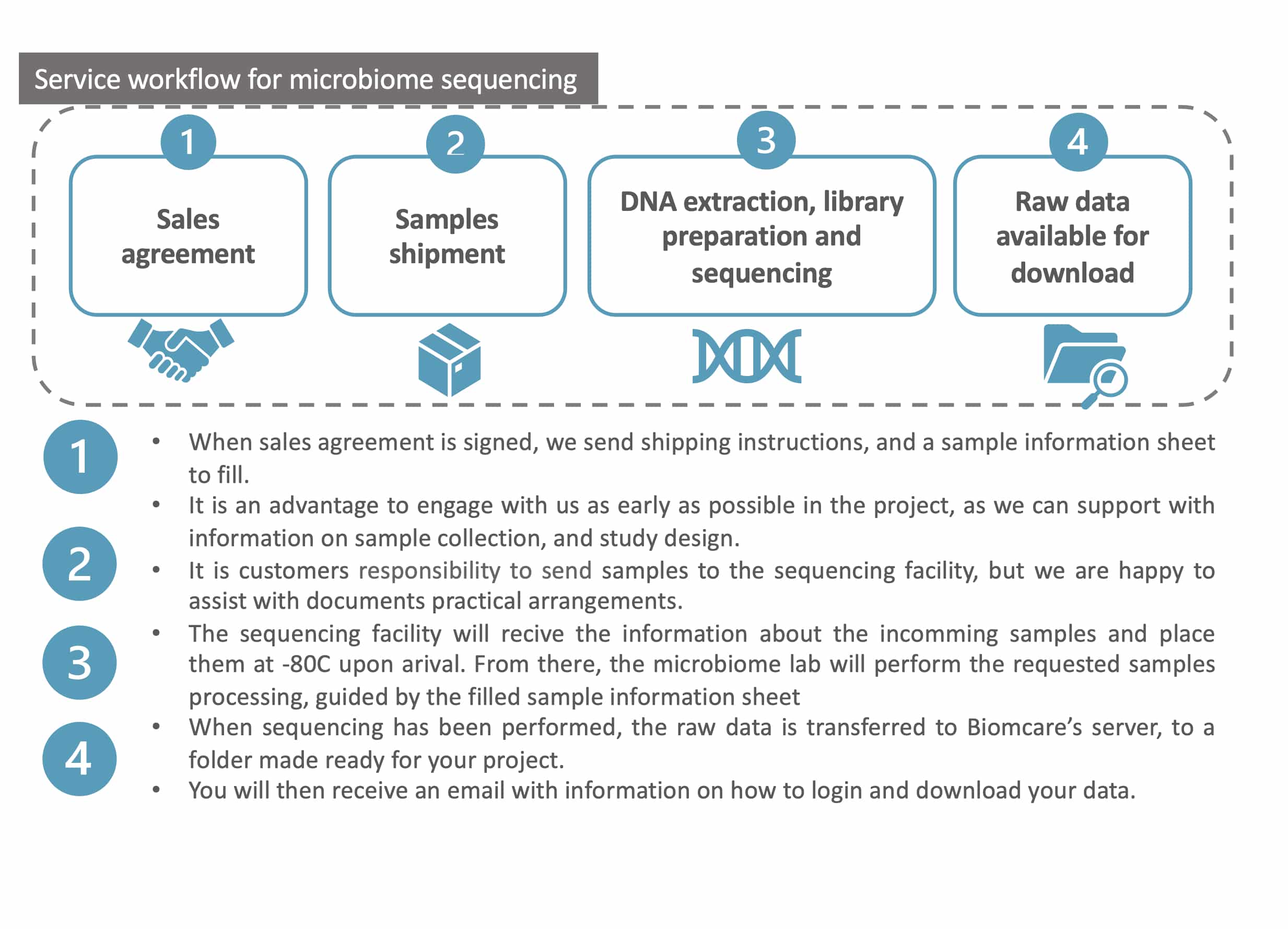
What we do
- Support with study design and guidance on sample collection and shipment.
- DNA extraction*
- Library preparation and amplicon sequencing on an Illumina platform
- If you purchase our biostatistical service, we also perform
- Data quality evaluation and quality filtering to prepare the data for microbiome profiling
- Microbiome profiling – calculation of the abundance of specific organisms (including absolute bacterial abundances if spike-in solutions are used*)
- Statistical analysis
* Please note the option to use spike-in solutions for the calculation of absolute abundance. By default, sequencing methods provide information about the relative abundance of microorganisms in a sample. By using spike-in of known concentrations of microorganisms, we can calculate the actual number of organisms in a sample.

Solving your microbiome problems
We have more than +7 years of experience in analyzing microbiome data and structuring microbiome projects. Our team has worked on more than 75 microbiome projects spanning research, universities and commercial industries.
Biomcare offers both microbiome services for small discovery projects, as well as large custom-designed microbiome projects.
Contact us today and get a quote. We are standing by to service you.
What you get

For each project, a private project folder is made available on the Biomcare server, where all resulting data, statistical results, illustrations, reports, etc. are available for download.
For projects where you have purchased both sequencing and data analysis, we deliver three reports with descriptions of the data, the processing, analysis, and the results of the statistical analysis, together with illustrations and tables.
Sample collection barcodes, tubes, or kits
- Depending on the type of sample you collect, we will send a table for filling in your sample information, or we will ship barcode stickers or barcode-labeled tubes. For larger clinical projects, we can provide collection kits if requested.
Data
- Raw sequencing data in fastq format
- Quality filtered data in fastq format
Results reports
Three reports are generated with all necessary information on sequencing, data processing, and statistical analysis, and all results, illustrations, and tables.
- A report describing the performed sequencing, data evaluation, and processing
- A report describing the generated microbiome profiles, and first microbiome evaluations
- A report describing the statistical analysis performed and the results, including illustrations and tables.
Personal guidance and communication
A key factor for the successful outcome of every project is communication. Every project is different and good personal communication allows us to understand exactly what our customers wish to achieve. This is especially essential when we design the statistical analysis and incorporate customer-provided information (referred to as meta-data). We typically have the most active communication at the beginning when samples are being collected and shipped, and then again when statistical analysis is initiated and results start to emerge. At the end of the project, we present the project results in an online or face-to-face meeting, depending on geographical circumstances and project size and complexity.
"We used Biomcare to analyze the occurrence and composition of bacteria and fungi in soil samples, in a project evaluating soil fertility and the effect of tilling.
...The results were presented in easily understandable and illustrative reports, and by personal communication, and all communication went swiftly. I can highly recommend Biomcare for their expertise."
About the 16S rRNA gene and ITS sequences

"We used Biomcare to analyze paired human faecal samples from a clinical trial. The communication was swift and the samples rapidly analysed.
… All in all, making the data easily accessible to also less experienced in the field of 16S sequencing."
Figure: The concept of amplicon sequencing here is illustrated for the variable region V1V2 of the 16S rRNA gene for bacterial profiling.
The 16S rRNA gene encodes a ribosomal subunit. The gene sequence comprises several conserved and variable regions. The conserved regions are consistent across bacteria and allow us to use a single set of primers to target the bacteria in a sample. We then sequence a subset of variable regions between the targeted conserved regions. By sequencing the variable regions, which are specific for each species of bacteria, we can identify which bacteria are present in a sample and calculate their relative abundance*. This is possible with a resolution down to the species level. We use the software DADA2 to identify bacterial representative sequences and then use a reference database to annotate the detected bacteria.
The ITS2 sequence is situated between two ribosomal RNA genes (5.8S and 28S) and like the variable regions in the 16S rRNA gene, has highly conserved flanking sequences that allow the binding of primers for targeted amplicon sequencing. Due to its high degree of variation between even closely related species, it is recommended as the universal barcode for fungi phylogenetic profiling. By sequencing the ITS2 sequence we can identify fungi-representative sequences in a sample and their relative abundance, and by mapping to reference databases we can annotate the identified organisms.
By applying our data processing and biostatistical platform, we can evaluate if a certain test condition, such as a new food ingredient, affects the overall composition or diversity of the microbiome, and identify the indicator microorganism(s).
* Please note the option to use spike-in solutions for the calculation of absolute abundance. By default, sequencing methods provide information about the relative abundance of microorganisms in a sample. By using spike-in of known concentrations of microorganisms, we can calculate the actual number of organisms in a sample.
Solving your microbiome problems
We have more than +7 years of experience in analyzing microbiome data and structuring microbiome projects. Our team has worked on more than 75 microbiome projects spanning research, universities and commercial industries.
Biomcare offers both microbiome services for small discovery projects, as well as large custom-designed microbiome projects.
Contact us today and get a quote. We are standing by to service you.